Termodinámica de la adsorción. Termodinámica de los procesos de adsorción.
La adsorción tiene lugar en la interfaz. Por tanto, es razonable considerar la descripción termodinámica de los fenómenos superficiales como un caso especial de la termodinámica de sistemas heterogéneos.
Arroz. 3.4. Adsorción de Gibbs: 1- sistema de comparación de dos fases, 2- sistema real de dos fases con una región no uniforme
En la termodinámica de sistemas heterogéneos se utiliza. principio de aditividad que es el siguiente: Todas las propiedades extensivas de un sistema heterogéneo son iguales a la suma de las correspondientes propiedades extensivas que habrían tenido las fases antes de ponerse en contacto. Denotemos las fases por α y β (Fig. 4). Entonces, para un sistema ideal, tal que las propiedades de las fases cercanas a la interfaz coincidan con sus propiedades generales, las siguientes relaciones son válidas para la energía interna U, volumen V, masa (número de moles) n, entropía S después de establecer el equilibrio en un sistema heterogéneo:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Esto supone que la temperatura y la presión en ambas fases son las mismas.
Para sistemas heterogéneos reales, la región de transición en el límite de dos fases contribuye adicionalmente a las propiedades extensivas del sistema. Si ocurren fenómenos superficiales, se debe tener en cuenta la diferencia entre las propiedades extensivas de un sistema heterogéneo real y las propiedades extensivas de un sistema modelo en el que los fenómenos superficiales están ausentes. Un sistema de este tipo se llama sistema de comparación. El sistema de comparación tiene los mismos parámetros intensivos (T, P, C i...) y el mismo volumen V que el sistema real (Fig. 4).
Desde un punto de vista termodinámico, se entiende por valor de adsorción G la cantidad excedente de sustancia n s, expresada en moles o gramos, que tiene un sistema heterogéneo real en comparación con el sistema de referencia, en relación con el área de la interfaz o con el área de la superficie. del adsorbente A. Se supone que el sistema de comparación tiene los mismos parámetros intensivos (T, P, C i) y el mismo volumen (V = V α + V β) que el sistema real (Fig. 4) .
Г = (n - n α - n β)/A = n s /A 3.11
El exceso de funciones termodinámicas de la región de transición de un sistema real (las denotamos por el índice s) se puede escribir como
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β etc.
Las mediciones experimentales de adsorción siempre dan la adsorción precisamente como un exceso del componente en el sistema real en comparación con el sistema de referencia elegido. Por ejemplo, cuando se adsorbe un gas en un adsorbente sólido o cuando se adsorben componentes en una fase sólida, para encontrar los valores de adsorción, determine el cambio en las concentraciones iniciales del adsorbato después del contacto de las fases α y β.
norte yo s = V(C yo o - C yo),
Dónde C.i.o.– concentración inicial del i-ésimo componente, C yo– concentración del i-ésimo componente después de establecer el equilibrio entre las fases en contacto. Se cree que el volumen V no cambia. Sin embargo, la concentración iº componente C yo, obtenido experimentalmente, se determina en volumen V' por encima de la interfaz de fase sin tener en cuenta el volumen de la región no homogénea de la capa de transición V α en la interfaz donde la concentración es C i α. Así, debido a la existencia de una región no uniforme en un sistema real, el volumen total del sistema se puede representar como V = V’ + Vα. Toda cantidad i-ésimo componente C.i.o. se distribuirá entre estos dos volúmenes:
V C i o = V’ C i + V α C i α ,
y el número de moles del componente i, adsorbido en la interfaz, será igual a
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Aquellos. La adsorción determinada experimentalmente es el exceso del i-ésimo componente en el volumen V α en comparación con la cantidad de este componente en el mismo volumen lejos de la interfaz de fase. Este tipo de adsorción se llama adsorción de Gibbs. .
V α C yo α llamado contenido completo i-º componente de la capa de adsorción. En la región de concentraciones muy bajas C yo en volumen V' enmienda VαCi La ecuación (3.2) puede despreciarse y el valor medido puede considerarse V α C yo α contenido completo i-º componente en la capa de adsorción, por ejemplo, durante la adsorción de gas sobre un adsorbente sólido a bajas presiones.
En el caso de interacción entre dos átomos:
U – energía de interacción;
U = U ANTERIOR. + U VOLVER
 - Ecuación de Lennard-Jones
, c, b, m = constante
- Ecuación de Lennard-Jones
, c, b, m = constante
En los casos de interacción de átomos con una superficie sólida, es necesario sumar todas las interacciones.
x – distancia a la superficie
r – radio de acción de las fuerzas de atracción
dV – volumen
n – número de moléculas de superficie
U ADS. – energía de interacción de adsorción



En el caso de la adsorción, la atracción aumenta. Y en el caso de la interacción no polar-no polar, la adsorción se localiza predominantemente en las hendiduras.
Interacción electrostática.
Adsorbente polar – adsorbato no polar
Adsorbente no polar – adsorbato polar
Adsorbente polar – adsorbato polar.
METRO  La molécula de adsorbato se representa como un dipolo y el adsorbente se representa como un conductor en el que la molécula de adsorbato induce un espejo dipolar simétricamente con respecto al dado.
La molécula de adsorbato se representa como un dipolo y el adsorbente se representa como un conductor en el que la molécula de adsorbato induce un espejo dipolar simétricamente con respecto al dado.
X – distancia al medio
Al interactuar surge el potencial:
 ,
,
 - momento bipolar.
- momento bipolar.
El potencial tiende a tomar el valor máximo, es decir los dipolos tienden a orientarse perpendicularmente a la superficie.
Dado que un aumento de temperatura promueve el crecimiento del movimiento browniano, conduce a la inhibición del proceso de adsorción.
En el caso de interacción electrostática, el adsorbato se localiza predominantemente en las protuberancias.
Ecuación fundamental de adsorción.
En el caso de la adsorción se produce una redistribución del componente, lo que significa que cambia el potencial químico. El proceso de adsorción puede considerarse como la transición de energía superficial a energía química.
Volumen de capa = 0, entonces la ecuación generalizada de las leyes I y II de la termodinámica:
T = constante; (1) = (2) => 

Para un sistema de dos componentes:
,
 ,
,
 =>
=>
=>
 - Ecuación de adsorción de Gibbs
.
- Ecuación de adsorción de Gibbs
.
Para el caso de adsorción de TV. cuerpo - gas: , 
 ,
,

 - isoterma
- isoterma
 - isobara
- isobara
 - isopicnal
- isopicnal
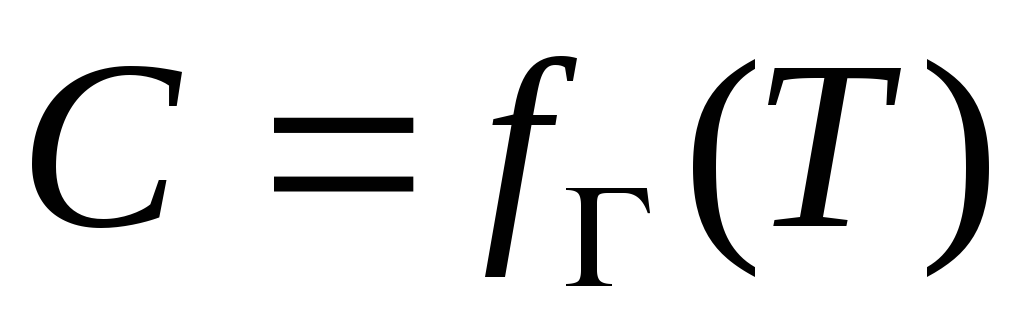 - isóstero
- isóstero
Isoterma, isopycne, isostere están relacionados entre sí.
Porque función de adsorción 



Isoterma de Henry Isoterma de Langmuir



Termodinámica. Adsorción.
Para materia condensada:
 ,
,
 ,
,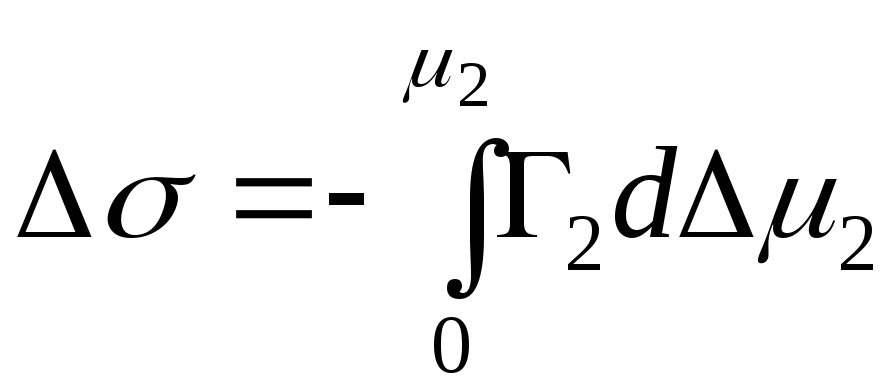
 - cambio integral en la energía de Gibbs
.
- cambio integral en la energía de Gibbs
.

P – presión sobre una superficie curva, Р S – presión sobre una superficie plana
 - potencial de adsorción
- potencial de adsorción
Cambio diferencial en la trampa. 
 , Г = constante
, Г = constante
- cambio de entropía diferencial
- entalpía diferencial de adsorción
 - calor isostérico de adsorción
- calor isostérico de adsorción
 - calor de condensación
- calor de condensación
 - calor neto de adsorción
- calor neto de adsorción

 ,
,




Qa – calor integral de adsorción, 
Qra – calor neto integral de adsorción,

la ecuación de henry
El estudio de la adsorción se complica por la heterogeneidad de la superficie, por lo que las leyes más simples se obtienen para superficies homogéneas.
Consideremos la interacción de los gases con una superficie sólida, cuando un gas pasa de un estado de equilibrio en el volumen a un estado de equilibrio en la superficie. Este caso es análogo al equilibrio de gases en un campo de gravedad.
 ,
,
 ,
=>
,
=> -la ecuación de henry
-la ecuación de henry
 - Coeficiente de distribución
- Coeficiente de distribución
Durante el proceso de adsorción, se produce un cambio en los potenciales químicos.
Para la fase masiva: 
Para gas en la superficie: 
En estado de equilibrio  , es decir.
, es decir.

En la ecuación de Henry la constante no depende de la concentración.
La ecuación de Henry es válida en la región de bajas presiones y concentraciones. A medida que aumenta la concentración, son posibles dos tipos de desviaciones de la ley de Henry:
1 – desviaciones positivas, D disminuye, A disminuye
2 – desviaciones negativas, D – aumentos, A – aumentos.
El tipo de desviación está determinado por el predominio de uno u otro tipo de interacción adsorbente-adsorbato.
Con una fuerte interacción adhesiva, los coeficientes de actividad aumentan: una desviación positiva. En el caso de interacciones cohesivas se observan desviaciones negativas.
Adsorción monomolecular.
Isoterma de Langmuir.
Los patrones más simples se obtuvieron en la teoría de Henry. Langmuir propuso una teoría según la cual la adsorción se considera una reacción cuasiquímica. Donde:
La superficie es energéticamente homogénea.
La adsorción es localizada, cada centro de adsorción interactúa con una molécula de adsorbato.
Las moléculas de adsorbato no interactúan entre sí.
Adsorción monocapa.

 - superficie,
- superficie,  - adsorbato,
- adsorbato,  - complejo de adsorción.
- complejo de adsorción.
 , entonces la concentración de sitios de adsorción:
, entonces la concentración de sitios de adsorción:  ,
, - limitar la adsorción.
- limitar la adsorción.
 , entonces la constante de reacción es:
, entonces la constante de reacción es: 

 - Ecuación de Langmuir.
- Ecuación de Langmuir.
Dependencia de la adsorción de la concentración.
1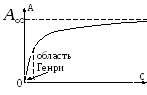 )
)
 ,
,

2) zona de altas concentraciones
 - limitación de la adsorción, formación de una capa monomolecular
- limitación de la adsorción, formación de una capa monomolecular
Para la energía de Gibbs: .
g es el factor de entropía.
En el caso de la isoterma de Henry, la energía de Gibbs caracteriza la transición del adsorbato del estado estándar en la masa al estado estándar en la superficie. En el caso de la isoterma de Langmuir  Caracteriza el grado de afinidad entre el adsorbente y el adsorbato.
Caracteriza el grado de afinidad entre el adsorbente y el adsorbato.
 encontrado en la isobara de van't Hoff.
encontrado en la isobara de van't Hoff.

 , Entonces
, Entonces  , de aquí
, de aquí  .
.

 - grado de relleno de la superficie.
- grado de relleno de la superficie.




 - número de asientos libres,
- número de asientos libres,  - número de plazas ocupadas.
- número de plazas ocupadas.
 ,
,

Aquellos. en la región de altas concentraciones, el número de sitios libres es inversamente proporcional a la cantidad de adsorbato.
Adsorción de una mezcla de gases sobre una superficie homogénea.
En este caso, el proceso de adsorción se considera como dos reacciones paralelas.
 (1)
(1)

 (2)
(2)


Adsorción de una mezcla de gases sobre una superficie no uniforme.
En el caso de una superficie no uniforme no se puede limitarse a empastes medios.
Como resultado de la competencia, es posible la localización de diferentes adsorbatos en áreas de diferentes tipos.
En este caso la relación  .
.
 ,
,
 - presión de vapor saturado del adsorbato.
- presión de vapor saturado del adsorbato.
 ,
,
 - calor de adsorción.
- calor de adsorción.
“+” - dependencia de simbat, “-” - dependencia de antibate, “N” - sin correlación.
“+” - la adsorción se produce según el mismo mecanismo. En las zonas energéticamente más favorables se adsorbe predominantemente gas con una alta afinidad por la superficie.
“-” - la adsorción se produce a través de varios mecanismos y hasta cierto punto no hay competencia por la superficie.
La adsorción monomolecular se realiza predominantemente durante la adsorción física de gases a valores bajos. pag, así como en la interfaz líquido/gas.
Adsorción polimolecular.
teoría APUESTA(Brunauer, Emmett, Teller).
En el caso en que la formación de una monocapa no sea suficiente para compensar la energía superficial, la adsorción es polimolecular y puede considerarse como resultado de la condensación forzada bajo la acción de fuerzas superficiales.
Puntos clave:
Cuando una molécula de adsorbato llega a un sitio ocupado, se forma un conjunto múltiple.
A medida que nos acercamos pag A pag s el número de sitios de adsorción libres disminuye. Inicialmente, el número de plazas ocupadas por individuales, dobles, etc. aumenta y luego disminuye. en conjuntos.
En pag =pag s la adsorción se convierte en condensación.
No hay interacciones horizontales.
Para la primera capa se cumple la isoterma de Langmuir.
La superficie se considera como un conjunto de sitios de adsorción. La condición de equilibrio dinámico es válida: la tasa de condensación en los lugares libres es igual a la tasa de evaporación en los lugares ocupados.
a es el coeficiente de condensación (la fracción de moléculas condensadas en la superficie);
 ,
,
Zm – número máximo de asientos libres.
 - frecuencia de las vibraciones atómicas en dirección perpendicular a la superficie.
- frecuencia de las vibraciones atómicas en dirección perpendicular a la superficie.
Para la primera capa, condiciones de equilibrio dinámico:



 , Entonces
, Entonces
 - Ecuación de Langmuir.
- Ecuación de Langmuir.
Para la segunda capa será cierto: 
Para la i-ésima capa: 
Por simplicidad, se supone que a y ν son iguales para todas las capas excepto la primera. Para todas las capas excepto la primera, el calor de adsorción es constante. Para la última capa, el calor de adsorción es igual al calor de condensación. Como resultado se obtuvo la ecuación
 (*)
(*)
C- constante,
En el caso de la teoría BET, la constante CON caracteriza la energía de Gibbs de adsorción pura. La ecuación contiene solo una constante y esta ecuación también es muy importante para determinar el área de superficie específica del adsorbente.
Dado que como resultado de la adsorción se libera calor, las superficies específicas se determinan a bajas temperaturas.
 ????????????
????????????


El principal inconveniente de la teoría.– descuidar las interacciones horizontales en favor de las verticales.
La ecuación se mantiene en el rango  de 0,05 a 0,3.
de 0,05 a 0,3.
Dónde  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – la interacción adsorbato-adsorbato se ve afectada.
> 0,3 – la interacción adsorbato-adsorbato se ve afectada.
Contabilización de interacciones adsorbato-adsorbato.
Las interacciones ocurren cuando moléculas o moléculas ramificadas se adsorben en una superficie no polar. Capaz de formar asociados. En este caso, cambia la forma de las isotermas de adsorción.
A  el adsorbente no es polar.
el adsorbente no es polar.
El gráfico 1 corresponde a interacciones adsorbato-adsorbato débiles y a interacciones adsorbato-adsorbente fuertes.
El gráfico 2 corresponde a interacciones fuertes adsorbato-adsorbato y fuertes adsorbato-adsorbente.
El gráfico 3 corresponde a la interacción adsorbato-adsorbato fuerte y a la interacción adsorbato-adsorbente débil.
 ,
,

En el caso de interacción entre moléculas de adsorbato, es necesario tener en cuenta los cambios en los coeficientes de actividad. Y esta ecuación se escribe como:
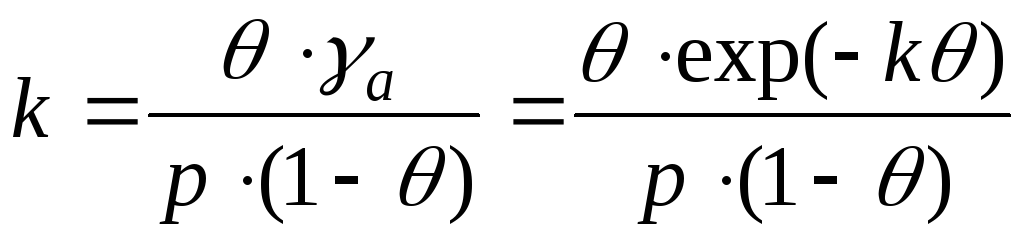 - Frunkin, Fowler, ecuación de Guggenheim.
- Frunkin, Fowler, ecuación de Guggenheim.
k– constante de atracción.
La teoría del potencial de Polyany.
Esta teoría no deriva ningún tipo de isoterma de adsorción, pero permite calcular isotermas a diferentes temperaturas.
Adsorción- este es el resultado de la atracción del adsorbato hacia la superficie del adsorbente debido a la acción del potencial de adsorción, que no depende de la presencia de otras moléculas y depende de la distancia entre la superficie y la molécula de adsorbato.
 ,
,
 - potencial de adsorción.
- potencial de adsorción.
Como la superficie no es uniforme, la distancia se reemplaza por el volumen de adsorción.  .Volumen de adsorción es el volumen encerrado entre la superficie y el punto correspondiente a un valor dado
.Volumen de adsorción es el volumen encerrado entre la superficie y el punto correspondiente a un valor dado  .
.
Potencial de adsorción es el trabajo de transferir 1 mol de adsorbato fuera de un volumen de adsorción dado a un punto dado del volumen de adsorción (o el trabajo de transferir 1 mol de vapor saturado de un adsorbato que está en equilibrio con un adsorbato líquido en ausencia de un adsorbente en fase de vapor en equilibrio con el adsorbente).

Curva característica
 - potencial de adsorción,
- potencial de adsorción, 
Para un adsorbente determinado y varios adsorbatos, se cumple lo siguiente: 
Para diferentes tipos de adsorbatos  ,
,
Dónde  Potenciales para isotermas de adsorción a presiones relativas.
Potenciales para isotermas de adsorción a presiones relativas.  para el adsorbato 1 y para el adsorbato 2. Esta relación es un valor constante.
para el adsorbato 1 y para el adsorbato 2. Esta relación es un valor constante.
 - coeficiente de afinidad
- coeficiente de afinidad
Teoría de la condensación capilar.
El curso del proceso de adsorción depende en gran medida de la estructura del cuerpo poroso.
|
microporoso | |
|
Poroso de transición | |
|
Macroporoso |
En el caso de los sorbentes microporosos, los campos de fuerzas de adsorción se superponen. En el caso de los sorbentes macroporosos, los poros actúan como canales de transporte. Los procesos de condensación son más significativos en cuerpos transicionalmente porosos. La condensación capilar comienza a ciertos valores. pag Y  , cuando ya se ha compensado parte de la energía superficial. Una condición necesaria es que la superficie sea autohumectante. El proceso se describe Ecuación de Thompson-Kelvin.
, cuando ya se ha compensado parte de la energía superficial. Una condición necesaria es que la superficie sea autohumectante. El proceso se describe Ecuación de Thompson-Kelvin.

 - para el caso de humectación, el centro de curvatura está en la fase gaseosa.
- para el caso de humectación, el centro de curvatura está en la fase gaseosa.
En el caso de la condensación capilar, la isoterma de adsorción tiene una forma histerética. La rama inferior corresponde al proceso de adsorción y la rama superior corresponde al proceso de desorción.
Todo tipo de poros se pueden reducir a tres tipos:
|
Cónico |
Cilíndrico con un extremo cerrado |
Cilíndrico con dos extremos abiertos |
|
El proceso de llenado se realiza desde el fondo del poro. La isoterma de adsorción y la isoterma de desorción en este caso coinciden, ya que el proceso de adsorción comienza a partir de una esfera y el proceso de desorción también comienza con la desaparición de algunas esferas.
↓ |
No hay histéresis. La carrera de avance y retroceso se describe mediante la ecuación:
|
No hay fondo por ningún lado, el llenado de los poros irá a lo largo de las paredes del cilindro.
cilindro: La isoterma tendrá una apariencia histerética.
↓ |


 - esfera,
- esfera, ,
,



EN  En condiciones de humedad, la condensación se produce a presiones más bajas, lo que es energéticamente favorable. A partir de la rama de desorción se obtienen las curvas de distribución del tamaño de poro.
En condiciones de humedad, la condensación se produce a presiones más bajas, lo que es energéticamente favorable. A partir de la rama de desorción se obtienen las curvas de distribución del tamaño de poro.
El máximo de la curva diferencial se desplaza hacia la izquierda con respecto al punto de inflexión de la curva integral. El volumen total de poros pequeños es pequeño, pero tiene grandes superficies. Al aumentar el tamaño de los poros, su volumen aumenta a medida que  , y el área es como
, y el área es como  , debido a esto se observa un desplazamiento en el máximo de la curva diferencial.
, debido a esto se observa un desplazamiento en el máximo de la curva diferencial.
Adsorción en la interfaz sólido-líquido.
En el caso de la adsorción en la interfaz sólido-gas, descuidamos un componente. En el caso de la adsorción en la interfaz sólido-líquido, el adsorbato desplaza las moléculas de disolvente de la superficie del adsorbente.
 ,
,
La ecuación es correcta:

 ,
,
N 1, N 2 – fracciones molares de disolvente y componente, N 1 + N 2 = 1, entonces
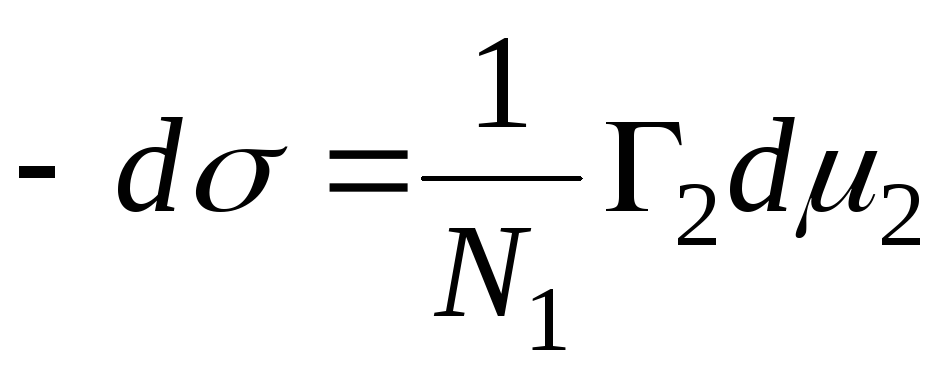 ,
=>
,
=>
 , entonces es la ecuación de adsorción para la interfaz sólido-líquido.
, entonces es la ecuación de adsorción para la interfaz sólido-líquido.
Adsorción (G) > 0 en  <
0
<
0
Si los valores  para el componente y el disolvente son muy diferentes, en este caso la dependencia GRAMO de norte tiene un extremo en el valor norte
~ 0,5.
para el componente y el disolvente son muy diferentes, en este caso la dependencia GRAMO de norte tiene un extremo en el valor norte
~ 0,5.
mi  si
si  tienen valores cercanos, en este caso el signo de adsorción puede cambiar. Adiccion GRAMO de norte cruza el eje x
tienen valores cercanos, en este caso el signo de adsorción puede cambiar. Adiccion GRAMO de norte cruza el eje x
Punto de intersección de funciones GRAMO(norte) con el eje x se llama azeótropo de adsorción. Esto significa que los dos componentes no se pueden separar en un adsorbente determinado.
Ecuación de isoterma de adsorción con constante de intercambio.
Durante la adsorción en la interfaz sólido-líquido, se produce una redistribución constante de componentes entre la superficie del adsorbente y el volumen de la solución.
 - componentes (- - se refieren a la superficie)
- componentes (- - se refieren a la superficie)

 ,
,
 ,
, .
.
 ,
,


Adsorción en la interfaz líquido-gas.
R  Consideremos el cambio en el perfil de concentración cuando se cruza la interfaz líquido-gas. Sea volátil el componente 2.
Consideremos el cambio en el perfil de concentración cuando se cruza la interfaz líquido-gas. Sea volátil el componente 2.
Cs – concentración en la capa superficial.
Basado en la definición de exceso de adsorción.

Si el componente no es volátil, entonces el valor de adsorción se escribirá de la siguiente manera:
PAG  Rhode Island
Rhode Island 

En la ecuación.  la naturaleza de una sustancia se describe por su derivada
la naturaleza de una sustancia se describe por su derivada  .
.
La isoterma de tensión superficial puede ser de la forma 1 o 2:
1 – tensioactivos
2 – tensioactivos
La actividad superficial g es la capacidad de las sustancias para reducir la tensión superficial en un sistema.

 - espesor de la capa superficial
- espesor de la capa superficial
C s– concentración del componente en la capa superficial
CON– concentración de volumen
Para una serie homóloga existe una regla:
 - Regla de Traubo Duclos
- Regla de Traubo Duclos
Para una serie homóloga, la isoterma de adsorción se ve así:
En lugar de A escribimos G, ya que la adsorción es excesiva en la capa superficial.
Isoterma de tensión superficial:

 - tensión superficial de un disolvente puro.
- tensión superficial de un disolvente puro.
 - ecuación fundamental de adsorción;
- ecuación fundamental de adsorción;
 - Ecuación de Langmuir.
- Ecuación de Langmuir.
Resolvámoslos juntos:

- Ecuación de Shishkovsky.
B– constante para la serie homóloga.
A- al pasar de un homólogo a otro aumenta de 3 a 3,5 veces 
![]()
1 – zona de bajas concentraciones
![]()
2 – concentración media
3 – capa monomolecular
Los tensioactivos son moléculas difílicas, es decir. incluyen un grupo polar y un radical hidrocarbonado apolar.
o es la parte polar de la molécula.
| - parte no polar de la molécula.
En un disolvente polar, las moléculas de tensioactivo están orientadas de tal manera que la parte polar de la molécula mira hacia el disolvente y la parte no polar es empujada a la fase gaseosa.
En la ecuación de Shishkovsky  , es constante para la serie homológica.
, es constante para la serie homológica.
El efecto tensioactivo comienza a aparecer con norte>5. En concentraciones superiores a la concentración de la capa monomolecular, se produce micelización en soluciones tensioactivas.
micela– se llama un agregado de moléculas de tensioactivo anfifílico, cuyos radicales hidrocarbonados forman un núcleo y los grupos polares se convierten en la fase acuosa.
Masa micelar – masa micelar.
h  número de moléculas – número de agregación.
número de moléculas – número de agregación.
micelas esféricas
En el caso de la micelización, se establece el equilibrio en la solución.
CMC: concentración crítica de formación de micelas.
Dado que consideramos que la micela es una fase separada:
Para una serie homológica existe una ecuación empírica:
a– energía de disolución del grupo funcional.
b – incremento del potencial de adsorción, trabajo de adsorción por unidad de metileno.
– incremento del potencial de adsorción, trabajo de adsorción por unidad de metileno.
La presencia de un núcleo de hidrocarburo en las micelas crea la oportunidad para que compuestos que son insolubles en agua se disuelvan en soluciones acuosas de tensioactivos; este fenómeno se llama solubilización (lo que se disuelve es el solubilizado, el tensioactivo es el solubilizante).
El lodo puede ser completamente apolar, puede contener partes polares y no polares y estará orientado como una molécula de tensioactivo.
En cualquier caso, durante la solubilización se produce un aumento de la masa micelar y del número de agregación no sólo debido a la inclusión del solubilizado, sino también a un aumento del número de moléculas de tensioactivo necesarias para mantener un estado de equilibrio.
La solubilización es más eficaz cuanto menor es el peso molecular del solubilizado.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
La eficacia de los tensioactivos depende del valor de CMC.
Presión de la capa superficial 2D

→ -fuerzas de tensión superficial.
- presión bidimensional.
La capa superficial es una fuerza igual a la diferencia de tensión superficial de una solución tensioactiva y un disolvente puro, dirigida hacia una superficie limpia.
Se establece un equilibrio entre la solución y la capa superficial.



En  hay una zona donde
hay una zona donde  depende linealmente de la concentración.
depende linealmente de la concentración.
G [mol/m2].
 -área ocupada por un mol de una sustancia
-área ocupada por un mol de una sustancia
Entonces la isoterma de presión bidimensional tendrá la forma 
 - isoterma de presión bidimensional.
- isoterma de presión bidimensional.
Adiccion  de SM:
de SM:

En  - La presión bidimensional aumenta bruscamente. En
- La presión bidimensional aumenta bruscamente. En  bidimensional se deforma, provocando un crecimiento repentino
bidimensional se deforma, provocando un crecimiento repentino  .
.
Una película unida por fases idénticas en ambas caras se llama de doble cara. En tales películas se observa un movimiento constante de las aguas madre.
Las películas de menos de 5 nm de espesor se denominan películas negras.

Las capas de adsorción deben tener dos características: viscosidad y fácil movilidad, fluidez y elasticidad.
El efecto Marangoni es autocurativo.
triángulo de gibbs,  - sobrepresión.
- sobrepresión.
La película se ha estirado y, debido a que parte del líquido ha salido, los tensioactivos se precipitan hacia el espacio libre. Triángulo de Gibbs.
El efecto de la fuerza de adsorción de los cuerpos.
Siempre hay una capa de adsorción en la superficie de la película, por lo que luego
Ecuación de Langmuir:


 en presión bidimensional
en presión bidimensional
 - un análogo de la ecuación de Shishkovsky
- un análogo de la ecuación de Shishkovsky
Fenómenos electrocinéticos. Doble capa eléctrica (EDL).
Modelo Gelemholtz. Teoría de Gouy-Chapman.
Vuelo 1808

Ud. – tubo con forma, sumerja 2 electrodos en él. Se viola la ley de los vasos comunicantes y se produce un cambio en el nivel de líquido en el tubo: fenómenos electrocinéticos.
Fenómenos cinéticos:
Electroforesis
Electroósmosis
Potencial de flujo (flujo)
Potencial de sedimentación
1 y 2 surgen cuando se aplica una diferencia de potencial; 3 y 4, el punzonado y la sedimentación de partículas coloidales provocan la aparición de una diferencia de potencial.
Electroósmosis es el movimiento de un medio de dispersión con respecto a una fase dispersa estacionaria bajo la influencia de una corriente eléctrica.
Electroforesis – este es el movimiento de partículas en fase dispersa con respecto a un medio de dispersión estacionario bajo la influencia de una corriente eléctrica.
PAG  El motivo de la aparición de fenómenos electrocinéticos es la separación espacial de cargas y la aparición de una doble capa eléctrica.
El motivo de la aparición de fenómenos electrocinéticos es la separación espacial de cargas y la aparición de una doble capa eléctrica.
La doble capa eléctrica es un condensador plano; una placa está formada por iones que determinan el potencial y la otra por contraiones. Los iones se contaminan del mismo modo que los coiones que determinan el potencial se introducen en el volumen de la solución. Distancia entre placas  . El potencial cae linealmente, la diferencia de potencial
. El potencial cae linealmente, la diferencia de potencial  .
.
Una diferencia de potencial externa provoca la aparición de un módulo de corte.  es un par de fuerzas por unidad de área que actúan a lo largo de la superficie de un cuerpo sólido.
es un par de fuerzas por unidad de área que actúan a lo largo de la superficie de un cuerpo sólido.

En equilibrio, el módulo de corte es igual al módulo de fricción viscosa (  ).
).

en nuestras condiciones  ,
,

 - Ecuación de Gelemholtz-Smalukowski
- Ecuación de Gelemholtz-Smalukowski
 - velocidad lineal de desplazamiento de fase.
- velocidad lineal de desplazamiento de fase.
mi– intensidad del campo eléctrico.
 - diferencia de potencial entre placas
- diferencia de potencial entre placas
 - movilidad electroforética [m 2 /(V*s)].
- movilidad electroforética [m 2 /(V*s)].
El modelo de Helemholtz no tiene en cuenta el movimiento térmico de las moléculas. En realidad, la distribución de iones en la doble capa es más compleja.
Gui y Chapman identificaron las siguientes causas de DES:
La transición de un ion de una fase a otra cuando se establece el equilibrio.
Ionización de materia en fase sólida.
Finalización de la superficie con iones presentes en el medio de dispersión.
Polarización de una fuente de corriente externa.
La doble capa eléctrica tiene una estructura difusa o difusa. Los iones tienden a distribuirse uniformemente por toda la capa difusa.
La capa difusa está formada por contraiones; la longitud de la capa está determinada por su energía cinética. A temperaturas cercanas al cero absoluto, los contraiones están lo más cerca posible de los iones que determinan el potencial.
La teoría de Danya se basa en dos ecuaciones:
ecuación de Boltzmann

 - trabajar contra las fuerzas de interacción electrostática.
- trabajar contra las fuerzas de interacción electrostática.
 - densidad de carga volumétrica.
- densidad de carga volumétrica.

ecuación de poisson 
Dado que el espesor de la EDL es mucho menor que el tamaño de partícula y para una EDL plana la derivada con respecto a las coordenadas  Y
Y  está abolido.
está abolido. 
para e y en y<<1 функцию можно разложить в ряд Маклорена:

Limitémonos entonces a dos términos de la serie:


 - El espesor DEL es la distancia a la que el potencial DEL disminuye en mi una vez.
- El espesor DEL es la distancia a la que el potencial DEL disminuye en mi una vez.
Cuanto más baja es la temperatura, menos  . En T→0 – DEL plano. Cuanto mayor es la concentración, cuanto más yo, menos
. En T→0 – DEL plano. Cuanto mayor es la concentración, cuanto más yo, menos  .
.





 “–” significa que el potencial disminuye con la distancia. =>
“–” significa que el potencial disminuye con la distancia. =>
=>

 ,
,
 - el potencial disminuye exponencialmente.
- el potencial disminuye exponencialmente.
Potencial de densidad de carga superficial: 
La carga superficial es una carga volumétrica de signo opuesto, integrada a lo largo de la distancia.




=>


Donde el potencial disminuye 2,7 veces - 
Capacidad de doble capa 
La desventaja de la teoría es que no se tiene en cuenta la presencia de la capa de Helemholtz, es decir, no tiene en cuenta  , de ahí los errores en la determinación de los principales parámetros. Tampoco explica la influencia de iones de diferente naturaleza sobre el espesor de la doble capa eléctrica.
, de ahí los errores en la determinación de los principales parámetros. Tampoco explica la influencia de iones de diferente naturaleza sobre el espesor de la doble capa eléctrica.
La teoría de Stern. Estructura de una micela coloidal.
La doble capa eléctrica consta de dos partes: densa y difusa. Se forma una capa densa como resultado de la interacción de iones formadores de potencial con los adsorbidos específicamente. Estos iones suelen estar parcial o completamente deshidratados y pueden tener la misma carga o la carga opuesta a la de los iones que determinan el potencial. Depende de la proporción de energía de interacción electrostática.  y potencial de adsorción específico
y potencial de adsorción específico  . Los iones de la capa densa están fijos. La otra parte de los iones se encuentra en la capa difusa; estos iones están libres y pueden penetrar profundamente en la solución, es decir. desde una zona de mayor concentración a una zona de menor concentración. La densidad de carga total consta de dos partes.
. Los iones de la capa densa están fijos. La otra parte de los iones se encuentra en la capa difusa; estos iones están libres y pueden penetrar profundamente en la solución, es decir. desde una zona de mayor concentración a una zona de menor concentración. La densidad de carga total consta de dos partes.

 -carga de la capa de Helmholtz
-carga de la capa de Helmholtz
 -Carga de capa difusa
-Carga de capa difusa
La superficie tiene un cierto número de centros de adsorción, cada uno de los cuales interactúa con un contraión. La constante de tal reacción cuasiquímica es igual a:

 , Dónde
, Dónde  - fracción molar de contraiones en solución
- fracción molar de contraiones en solución
Distribución de Helmholtz
El potencial disminuye linealmente.
Distribución potencial de Gouy. No hay una capa densa, el potencial disminuye exponencialmente desde el valor 
Distribución popa.
Inicialmente, la disminución potencial es lineal y luego exponencial.
Cuando se aplica un campo eléctrico en el caso de la electroforesis, no es la partícula de la fase sólida la que se mueve directamente, sino la partícula de la fase sólida con una capa de iones rodeándola. El DES repite la forma de la partícula de fase dispersa. Cuando se aplica un potencial, se arranca parte de la capa difusa. La línea de ruptura se llama límite deslizante.
El potencial que surge en el límite de deslizamiento como resultado de la separación de parte de la capa difusa se llama potencial electrocinético(Potencial zeta  ).
).
Una partícula en fase dispersa con una capa circundante de contraiones y una doble capa eléctrica se llama micela.
Reglas para escribir micelas coloidales:


1-1 electrolito de carga
T – partícula en fase dispersa.
AA es el límite entre las partes densa y difusa.
BB – límite deslizante.
El límite deslizante puede coincidir o no con la línea AA.
El valor de pH en el que el potencial zeta es cero se llama punto isoeléctrico.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. Exceso de CaCl 2
CaCl2 ↔ Ca2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( norte-x)Cl - ) 2 X + X Cl - - notación micelar.
CaSO 4 m – agregado.
CaSO 4 m∙nCa 2+ – núcleo.
CaSO 4 m∙nCa 2+ 2( norte-x)Cl - - partícula.
2. Exceso de Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micela
CaSO 4 m – agregado.
CaSO 4 m∙nSO 4 2 + – núcleo.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - partícula
Ecuación de Gelemholtz-Smoluchowski

 - velocidad lineal de desplazamiento de límites (en electroósmosis).
- velocidad lineal de desplazamiento de límites (en electroósmosis).
 - diferencia de potencial entre las placas del condensador (en electroósmosis).
- diferencia de potencial entre las placas del condensador (en electroósmosis).


 - caudal volumétrico de la solución, S– área de la sección transversal de la celda.
- caudal volumétrico de la solución, S– área de la sección transversal de la celda.

mi– intensidad del campo eléctrico.


 (para electroósmosis).
(para electroósmosis).
Para potencial de flujo:

 - potencial
- potencial
 - presión sobre la membrana
- presión sobre la membrana
Como regla general, los valores de las movilidades electroforéticas y electroosmóticas son menores que los calculados. Esto sucede debido a:
Efecto de relajación (cuando una partícula en fase dispersa se mueve, se rompe la simetría de la atmósfera iónica).
Inhibición electroforética (la aparición de fricción adicional como resultado del movimiento de los contraiones).
Distorsión de las líneas de corriente en el caso de partículas eléctricamente conductoras.
Relación entre tensión superficial y potencial. Ecuación de Lippmann.
La formación de EDL se produce de forma espontánea debido al deseo del sistema de reducir su energía superficial. En condiciones de constante t Y pag la ecuación generalizada de la primera y segunda leyes de la termodinámica es la siguiente:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1ª ecuación de Lippmann.
- 1ª ecuación de Lippmann.
 - densidad de carga superficial.
- densidad de carga superficial.
 - capacitancia diferencial.
- capacitancia diferencial.
 - 2ª ecuación de Lippmann.
- 2ª ecuación de Lippmann.
CON- capacidad.
Resolvamos la primera ecuación de Lippmann y la ecuación fundamental de adsorción:
 ,
,


 , Entonces
, Entonces
 - Ecuación de Nernst
- Ecuación de Nernst
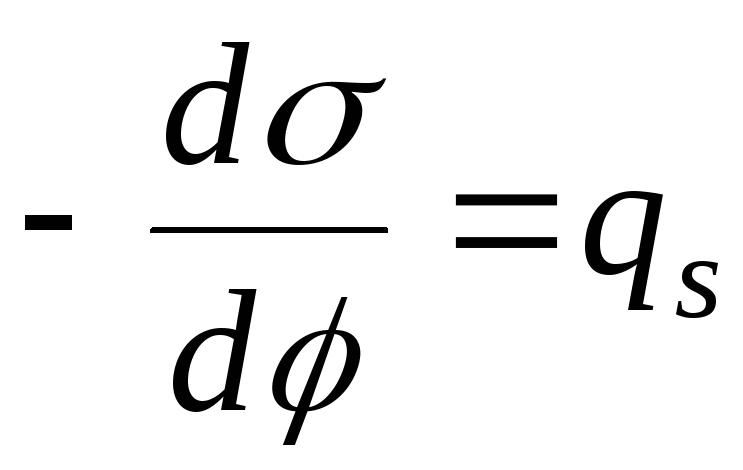 ,
,
 ,
,




 - ecuación de la curva electrocapilar (ECC).
- ecuación de la curva electrocapilar (ECC).
EN  :
: , Pero
, Pero 
Los tensioactivos catiónicos (CPAS) reducen la rama catódica del EKC.
Los tensioactivos aniónicos (APS) reducen la rama anódica del EKC.
Los tensioactivos no iónicos (NSAS) reducen la parte media de la CEC.
Estabilidad de sistemas dispersos. Presión desunión.
Los sistemas dispersos se pueden dividir:
Los sistemas que son termodinámicamente inestables pueden ser cinéticamente estables debido a la transición a un estado metaestable.
Hay dos tipos de estabilidad:
Estabilidad de la sedimentación (en relación con la gravedad).
Estabilidad agregativa. (relativo a la adhesión)
Coagulación Es un proceso de adhesión de partículas, que conduce a la pérdida de estabilidad de agregación. La coagulación puede ser causada por cambios de temperatura, pH, agitación y ultrasonido.
Se distingue la coagulación:
Reversible.
Irreversible.
La coagulación se produce con la introducción de electrolitos.
Reglas de coagulación:

Película- esta es la parte del sistema ubicada entre dos superficies interfaciales.
Presión desunión Ocurre cuando el espesor de la película disminuye drásticamente como resultado de la interacción de las capas superficiales que se aproximan.

“-” - a medida que disminuye el espesor de la película, aumenta la presión de separación.
P 0 es la presión en la fase masiva, que es una continuación de la capa intermedia.
P 1 – presión en la película.
Teoría de la estabilidad. DLFO (Deryagin, Landau, Calle, Overbeck).
Según la teoría DLFO, la presión de separación tiene dos componentes:
Electrostático P E (positivo, se debe a las fuerzas de repulsión electrostática). Corresponde a una disminución de la energía de Gibbs al aumentar el espesor de la película.
Molecular P M (negativo, debido a la acción de fuerzas de atracción). Es causada por la compresión de la película debido a fuerzas químicas superficiales, el radio de acción de las fuerzas es décimas de nm con una energía de aproximadamente 400 kJ/mol.
Energía de interacción total:

 - el sistema es agregativamente estable
- el sistema es agregativamente estable
 - sistema inestable
- sistema inestable
PAG  componente positivo.
componente positivo.
El aumento se debe a un aumento de la energía potencial cuando se comprimen películas delgadas. Para películas de gran espesor, el exceso de energía iónica se compensa y es igual a la interacción energética en el volumen del medio de dispersión.
Si  (
( - espesor de la película,
- espesor de la película,  - radio iónico) el adelgazamiento de la película conduce a la desaparición y reducción de moléculas e iones con una energía superficial mínima en ella. El número de partículas vecinas disminuye, como resultado de lo cual aumenta la energía potencial de las partículas que quedan en la película.
- radio iónico) el adelgazamiento de la película conduce a la desaparición y reducción de moléculas e iones con una energía superficial mínima en ella. El número de partículas vecinas disminuye, como resultado de lo cual aumenta la energía potencial de las partículas que quedan en la película.


La teoría DLVO considera la interacción de partículas como la interacción de placas.

Las partículas no interactúan.



 - ecuación de Laplace,
- ecuación de Laplace,  ,
,


Para superficies débilmente cargadas
Para superficies muy cargadas:

El componente molecular es la interacción de dos átomos:
 ~
~
Interacción de un átomo con una superficie:

Tomemos dos registros:
D  Para obtener el componente molecular, es necesario sumar todas las energías de interacción de los átomos de las placas derecha e izquierda.
Para obtener el componente molecular, es necesario sumar todas las energías de interacción de los átomos de las placas derecha e izquierda.
Dónde  - Constante de Hamaker (tiene en cuenta la naturaleza de los cuerpos que interactúan).
- Constante de Hamaker (tiene en cuenta la naturaleza de los cuerpos que interactúan).
Eso. La energía de interacción de las partículas en un sistema se puede expresar mediante curvas de potencial.
I – mínimo potencial primario. Esta es una zona de coagulación irreversible, prevalecen las fuerzas de atracción.
II – zona de estabilidad agregativa, predominan las fuerzas repulsivas.
III – mínimo de potencial secundario (o zona de floculación). Hay una capa de electrolito entre las partículas de la fase dispersa y las partículas se pueden separar y transferir a la zona de estabilidad de agregación.

Curva 1: el sistema es agregativamente estable.
Curva 2 – estable en la zona I, inestable en la zona II.
Curva 3: se ha producido coagulación en el sistema.
Curva 4 – en el punto 4 la energía de interacción total U=0,  , este punto extremo corresponde al inicio de la coagulación rápida.
, este punto extremo corresponde al inicio de la coagulación rápida.
Hay dos casos:
1. Superficies ligeramente cargadas:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - este es el espesor de la capa correspondiente al inicio del proceso de coagulación.
- este es el espesor de la capa correspondiente al inicio del proceso de coagulación.






 - para superficies débilmente cargadas
- para superficies débilmente cargadas
 Entonces
Entonces 
2. Para superficies muy cargadas:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Cuadremos (3)

Coagulación:
En la adsorción específica, los iones se pueden adsorber en cantidades superequivalentes de modo que la superficie pueda cambiar su carga. La superficie está recargada.
En el caso de una adsorción específica, no sólo se pueden adsorber iones de signos opuestos, sino también del mismo signo.
Si se adsorben iones del mismo signo que la superficie, entonces en la capa superficial no habrá una caída del potencial, sino un aumento.

Coagulación de neutralización (ocurre con la participación de partículas débilmente cargadas y depende no solo de la carga del electrolito coagulador, sino también del potencial en el límite de las capas densas y difusas).
Teoría de la coagulación rápida de Smoluchowski.

Dependencia de la velocidad de coagulación de la concentración de electrolitos.
I – la tasa de coagulación es baja,
II – la velocidad de coagulación es casi proporcional a la concentración de electrolitos.
III – región de coagulación rápida, la velocidad es prácticamente independiente de la concentración.
Disposiciones básicas:
El sol inicial es monodisperso, partículas similares tienen forma esférica.
Todas las colisiones de partículas son efectivas.
Cuando dos partículas primarias chocan, se forma una partícula secundaria. Secundaria + primaria = terciaria. Primaria, secundaria, terciaria – multiplicidad.
En términos de cinética química, el proceso de coagulación se puede describir mediante la ecuación:
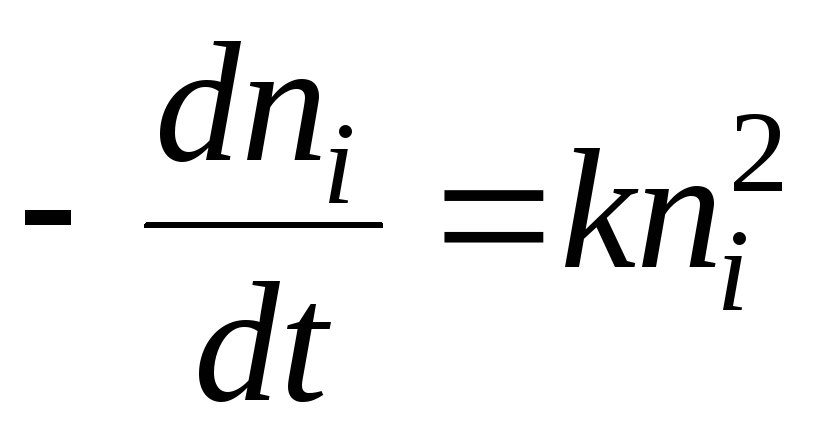
La solución será la ecuación: 
 - la mitad del tiempo de coagulación. Este es el tiempo durante el cual el número de partículas de sol se reduce 2 veces.
- la mitad del tiempo de coagulación. Este es el tiempo durante el cual el número de partículas de sol se reduce 2 veces.

 ,
,
 ,
,
 ,
,


A medida que aumenta la multiplicidad, el máximo de las curvas de coagulación se desplaza hacia valores mayores.  .
.
Defectos:
Supuesto de monodispersidad.
Supuesto sobre la efectividad de todas las colisiones.
Definiciones básicas y métodos de clasificación de procesos de adsorción.
La adsorción se refiere a fenómenos que ocurren debido a una disminución espontánea de la energía superficial.
Adsorción– el proceso de redistribución espontánea reversible o irreversible de los componentes de un sistema heterogéneo entre la capa superficial y el volumen de la fase homogénea.
En los sistemas multicomponente, el componente que reduce más fuertemente la tensión interfacial se transfiere preferiblemente a la capa superficial. En los sistemas monocomponente, durante la formación de la capa superficial, se produce un cambio en su estructura (una cierta orientación de átomos y moléculas, polarización), llamado autoadsorción.
La fase más densa en la que se localizan las interacciones de adsorción se llama adsorbente. La sustancia redistribuida entre el volumen de la fase homogénea y la capa superficial se indica con el término " adsorber».
En algunos casos, el proceso de adsorción es reversible. En este caso, bajo ciertas condiciones, parte de las moléculas adsorbidas, como resultado de fenómenos cinéticos moleculares, pueden pasar de la capa superficial a la fase masiva. El proceso inverso de adsorción se llama desorción.
Métodos de clasificación de procesos de adsorción.
Clasificación de los procesos de adsorción según el estado de agregación de las fases que interactúan. Dependiendo del estado de agregación de las fases adyacentes, se distinguen los siguientes tipos de procesos de adsorción:
Adsorción de gases sobre adsorbentes sólidos;
Adsorción de solutos en interfaces sólido-líquido y líquido-líquido;
Adsorción de tensioactivos en la interfaz líquido-gas.
Clasificación de los procesos de adsorción según el mecanismo de interacción entre el adsorbente y el adsorbato. La adsorción puede considerarse como la interacción de las moléculas de adsorbato con los centros activos del adsorbente. Según el mecanismo de su interacción, se dividen los siguientes tipos de adsorción:
1) adsorción física (molecular)– la interacción entre las moléculas del adsorbato y el adsorbente se realiza mediante fuerzas de van der Waals, enlaces de hidrógeno (sin reacciones químicas);
2) adsorción química (quimisorción)– la unión de las moléculas de adsorbato a los centros activos del adsorbente se produce como resultado de reacciones químicas de varios tipos (con excepción de las reacciones de intercambio iónico);
3) adsorción por intercambio iónico (intercambio iónico): redistribución de la sustancia adsorbida entre la solución y la fase sólida (intercambiador iónico) según el mecanismo de las reacciones de intercambio iónico.
Para describir cuantitativamente los procesos de adsorción, se utilizan dos cantidades.
1) Adsorción absoluta– cantidad (mol) o masa (kg) de adsorbato por unidad de superficie o masa del adsorbente. Designación – A; dimensión: mol/m2, mol/kg, kg/m2, kg/kg.
2) Adsorción de Gibbs (exceso)– exceso de sustancia adsorbato en una capa superficial de cierto espesor en comparación con su cantidad en el volumen de la fase homogénea, por unidad de superficie o masa del adsorbente. Designación – G; dimensión: mol/m2, mol/kg.
La relación entre la adsorción absoluta y el exceso se puede ilustrar mediante la ecuación:
Г = А – с * h (3.1)
donde с es la concentración de equilibrio de la sustancia en el volumen de la fase, mol/m3;
h es el espesor de la capa superficial, que convencionalmente se supone que es de 10 -9 m.
En sistemas heterogéneos multicomponentes, cuando uno u otro componente se redistribuye entre el volumen de la fase homogénea y la capa superficial, la ecuación para el exceso de energía interna de la superficie es válida:
U = T * S + s * s + Sm yo * n yo (3.2)
Reduciendo todos los términos de la ecuación a la unidad de área de la superficie de interfase, obtenemos:
U s = T * S s + s + Sm i * Г i (3.3)
donde Г i = n i / s es el exceso del i-ésimo componente en la capa superficial, es decir, la adsorción de Gibbs.
Para un sistema de un componente, la ecuación (3.3) tomará la forma:
G s = s + m * G (3.4)
donde G s = U s - T * S s – energía de Gibbs de la superficie o el trabajo de crear una unidad de superficie;
m * G – compactación de la sustancia de la sustancia adsorbida en la capa superficial.
Con base en la ecuación (3.4), podemos concluir que durante la adsorción, el trabajo de crear una superficie de interfase consiste en el trabajo de formación de la superficie (romper los enlaces cohesivos en el volumen de la fase de adsorbato) y la compactación de la sustancia en la capa superficial.
En un estado de equilibrio dinámico entre el adsorbente y el adsorbato, el cambio en la energía de Gibbs de un sistema heterogéneo ΔG = 0, la termodinámica del proceso de adsorción se describe mediante la ecuación denominada Ecuación fundamental de adsorción de Gibbs:
Ds = SГ i * dm i (3.5)
Esta ecuación es universal, ya que es válida para todo tipo de procesos de adsorción.
Casos especiales de la ecuación de adsorción de Gibbs.
1) Adsorción de soluciones.
Para el potencial químico del i-ésimo componente del sistema durante la adsorción en las interfaces “adsorbente líquido-sólido” y “líquido-gas”, son válidas las siguientes ecuaciones:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R*T* d ln a i (3.7)
donde m i 0 es el potencial químico del i-ésimo componente del sistema en condiciones estándar;
a i es la actividad del i-ésimo componente del sistema en condiciones estándar.
En base a esto, la ecuación de adsorción de Gibbs toma la forma:
Г i = - a i / R*T * (ds / da i) (3.8)
Para soluciones de no electrolitos tomamos a i = c i, entonces:
Г i = - с / R*T * (ds / dс) (3.9)
Para soluciones de electrolitos:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
donde с ± es la concentración iónica promedio de la solución;
n es el coeficiente estequiométrico.
2) Adsorción de sustancias de la fase gaseosa.
Según la ecuación de Mendeleev-Clayperon:
Р = ñ * R*T (3.11)
En este sentido, la ecuación de Gibbs para la adsorción de gases sobre adsorbentes sólidos se escribe de la siguiente forma:
Г i = - Р / R*T * (ds / dР) (3.12)
En la práctica, la ecuación de adsorción de Gibbs permite, basándose en mediciones de tensión superficial a varios valores de concentración de líquido o presión de gas de equilibrio, calcular la cantidad de adsorción de sustancias en la capa interfacial para la cual se determina la tensión superficial.
Kuznetsova E.S. y Buryak A.K. llevaron a cabo una comparación de las características termodinámicas de la adsorción de aminoácidos y sus asociados. El trabajo investigó la influencia de la estructura de los aminoácidos, sus dímeros y asociados con componentes eluyentes en su adsorción en la superficie de materiales de carbono. Se realizó un cálculo estadístico molecular de las características termodinámicas de adsorción (TCA) de aminoácidos aromáticos (fenilalanina, tirosina), aminoácido heterocíclico (triptófano) y sus dímeros con ácido trifluoroacético (TFA) sobre la superficie de carbón térmico grafitizado (GTS). afuera. Los datos obtenidos se comparan con los patrones de retención de aminoácidos en Hypercarb de carbón grafitizado poroso en condiciones de cromatografía líquida de alta resolución de fase reversa (RP HPLC). Se ha demostrado que los valores de retención de TCA y aminoácidos aumentan al aumentar la cadena de carbonos de estos compuestos.
Shkolin A.V. y Fomkin A.A. analizaron el comportamiento de las funciones termodinámicas (calor isostérico molar diferencial de adsorción, entropía, entalpía y capacidad calorífica) del sistema de adsorción AUK, adsorbente de carbono microporoso de metano, dependiendo de los parámetros de equilibrio de adsorción en el rango de temperatura de 177,65 a 177,65ºC. 393 K y presiones desde 1 Pa hasta 6 MPa. Tener en cuenta la influencia de la no idealidad de la fase gaseosa y la no inercia del adsorbente condujo a la aparición de una dependencia de la temperatura del calor isostérico de adsorción, especialmente en la región de altas presiones del adsorbente. Para el sistema en estudio, la principal influencia sobre las funciones termodinámicas del sistema de adsorción la ejerce la no idealidad de la fase gaseosa. La corrección por la falta de inercia del adsorbente en este rango de parámetros del sistema de adsorción no supera el 2,5%.
En el Instituto de Química General e Inorgánica de la Academia de Ciencias de la República de Uzbekistán Muminov S.Z. En su trabajo investigó los cambios en las propiedades superficiales y la estructura porosa de la montmorillonita al reemplazar los cationes intercambiables del mineral por polihidroxialuminio. El vacío térmico preliminar tiene un efecto significativo sobre las propiedades de adsorción de la montmorillonita de polihidroxialuminio en relación con el alcohol metílico. A partir de datos de una serie de isósteres de adsorción de CH3 sobre sodio deshidratado y montmorillonitas modificadas, medidos en un amplio rango de temperaturas, se estableció la dependencia del calor de adsorción de la cantidad de sustancia adsorbida.
NS Kazbanov, A.V. Matveeva y O.K. Krasilnikov realizó un estudio sobre la adsorción de fenol de soluciones acuosas mediante carbones activados como FAS, PAH y fieltros de carbón a temperaturas de 293, 313 y 343 K en el rango de concentración de 5 a 250 mmol/l. Se obtuvo mediante carbonización de polímeros a base de furfural una serie de muestras de FAS de carbón activado secuencialmente, caracterizado por una distribución estrecha del tamaño de poro. PAH es un carbón activado polimérico microporoso. El fieltro de carbono es un material fibroso a base de fibras de celulosa hidratadas. Los parámetros de la estructura porosa de los adsorbentes se determinaron a partir de isotermas de adsorción de vapor de nitrógeno a 77 K (ASAP-2020, Micromeritics, EE. UU.). Los estudios de adsorción de soluciones se realizaron mediante el método de ampolla en un termostato. Las muestras seleccionadas se analizaron mediante espectrofotometría. El análisis de las isotermas de adsorción en fase líquida obtenidas se realizó utilizando la teoría del llenado volumétrico de microporos (VFM) según la ecuación de Dubinin-Radushkevich (DR).
El efecto de la temperatura sobre la sorción de soluciones líquidas es ambiguo. Por un lado, en el caso de los adsorbentes microporosos, la penetración de moléculas en los poros de tamaño comparable a estas moléculas depende de la energía cinética y, en consecuencia, aumenta con la temperatura. Por otro lado, la adsorción física es un proceso exotérmico y la adsorción disminuye con la temperatura. La relación entre estos factores para cada sistema determina el curso de la dependencia de la adsorción con la temperatura.
La singularidad del sistema adsorbente-fenol es que tiene una dependencia inversa de la temperatura de las isotermas de adsorción porque A medida que la temperatura aumenta de 293 a 313 K, aumenta el valor límite de adsorción, lo que aparentemente se debe al efecto de tamiz molecular: al aumentar la temperatura, las moléculas de fenol pueden penetrar en poros más estrechos de los materiales de carbono. La adsorción ocurre principalmente en microporos, ya que los adsorbentes tienen una pequeña cantidad de mesoporos. A medida que aumenta el tamaño de los microporos, los valores máximos de adsorción aumentan significativamente, alcanzando 2,9 mmol/g para PAH, 8,5 mmol/g para FAS y 12,7 mmol/g para fieltro. Las isotermas de adsorción resultantes están bien descritas por la ecuación DR con un exponente igual a 2.
